海洋聚酮天然产物具有多种生物活性,并在新药开发中扮演着重要的角色。但是,海洋药物研发面临着样品采集困难、天然丰度低,结构复杂、合成难度大等诸多挑战。这些挑战所造成的药源匮乏成为了长期以来限制海洋药物研发的最大瓶颈。
宋振雷教授团队长期致力于海洋天然产物的高效化学合成研究。研究旨在打破海洋药物的药源瓶颈,从而推动后续更为深入和广泛的、具有自主知识产权的新药研发。近期,该团队基于所开发的特色硅试剂及反应方法,实现了海洋聚酮药物bryostatins,plocabulin的发散及规模化合成,以及海洋聚酮天然产物PM050463的首次和无保护基合成,相关进展如下。
进展一:海洋药物草苔虫素(bryostatins)的发散及克级规模合成
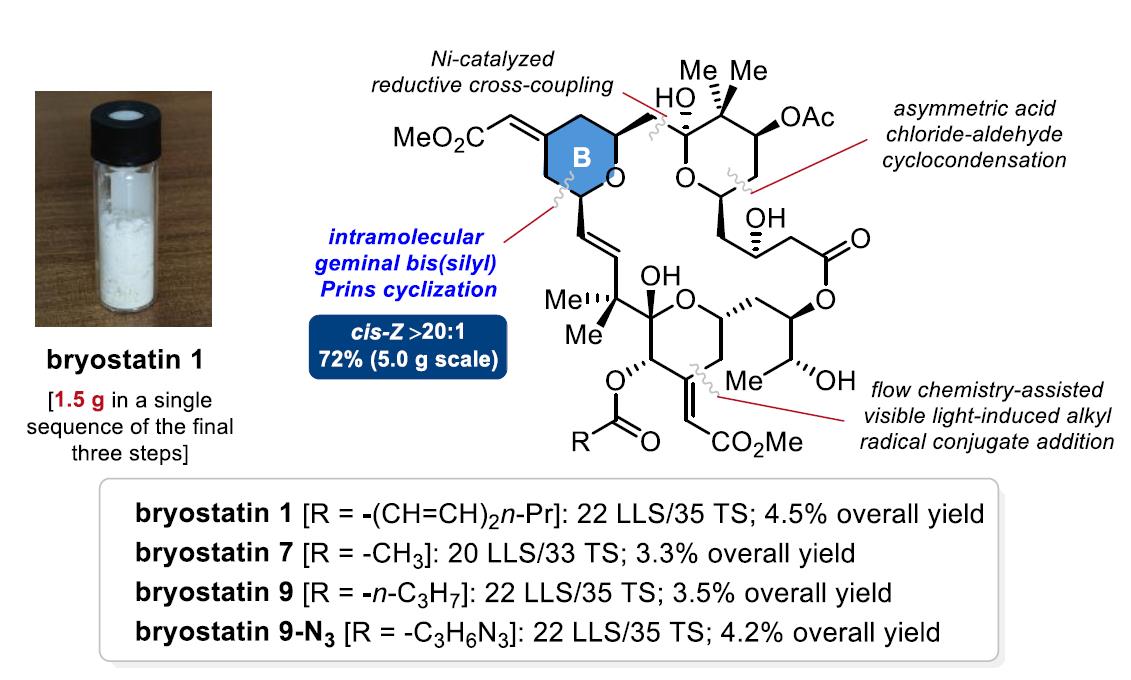
草苔虫素是一类具有高蛋白激酶C结合力(Ki=0.8-6 nM)的海洋大环内酯化合物,已开展阿尔兹海默等疾病共计44项临床试验。最新的中重度阿尔兹海默II期临床试验显示bryostatin-1疗效显著且安全性良好。但草苔虫素海洋采集困难,天然提取率不足百万分之一,药源极度稀缺。尽管自1990年代起全球有近20个团队针对其化学合成开展攻关,但因结构复杂、合成难度大,其单批次克级制备一直未被攻克。
该团队聚焦草苔虫素的化学合成开展了近15年的迭代攻关研究。2012年,应用所发展的偕二硅Prins环化,开发了B环一步构建的新方法(Angew. Chem. Int. Ed. 2012, 5367.);2015年,应用所发展的偕二硅远程Brook重排反应实现了C-环的高效构建(Chem. Commun 2013, 8961.);2018年,实现了草苔虫素-8的首次全合成(Angew. Chem. Int. Ed. 2018, 942.)。近期与秦勇教授团队合作所完成的第二代通用合成路线,不仅发散性地合成了4个成员分子(草苔虫素-1, 7, 9和9-N3,最长线性步骤20-22步/总步骤33-35步,总收率3.3-4.5%),并且在国际上率先实现了单批次克级制备(Angew. Chem. Int. Ed. 2025, e202423465),攻克了近30年的药物合成世界性难题,解决了草苔虫素的药源问题。根据阿尔兹海默 II 期临床试验单人用药量,首批制备的1.5 g 草苔虫素-1可满足约5000个患者的临床试验需求,并支撑更广泛的新适应症开发以及药物上市后的稳定供应。
相关成果以“热点文章”发表在Angew. Chem. Int. Ed. 2025,e202423465(https://onlinelibrary.wiley.com/doi/10.1002/anie.202423465)。研究得到了国家自然科学基金创新群体项目、国家重点研发计划等基金的资助,由我院博士生郭乾有、杨宇帆等共同完成。
进展二:海洋药物plocabulin及其类似物的发散及克级规模合成
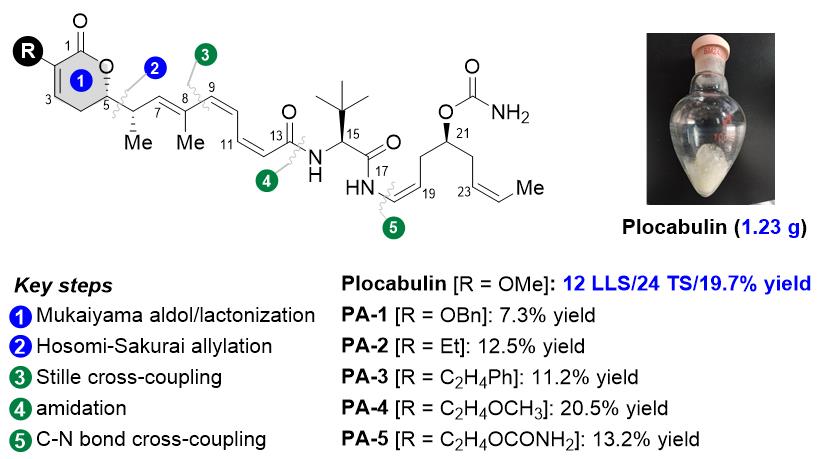
Plocabulin (PM060184)是一种源自马达加斯加海峡海绵的聚酮天然药物。研究表明,Plocabulin对微管蛋白的抑制作用呈现出一种新的作用机制,并在在特定细胞系中的活性较紫杉醇和长春碱高3-4个数量级,而且具有克服P-gp介导的多药耐药潜质。目前已开展5项临床研究,涉及转移性实体瘤、乳腺癌和晚期结直肠癌等适应症。但是,其天然丰度极低,仅有0.00003%。已知的2条合成路线步骤冗长、效率低下,很难实现规模化制备。因此,开发更加高效且适用于多样化制备的合成路线,对于打破其药源瓶颈,推动更加深入的成药性研究具有重要意义。
该团队创新性地发展了基于汇聚式合成策略的全新路线,成功实现Plocabulin的克级规模制备。合成的关键步骤包括:(1)TiCl4-螯合介导的Hosomi- Sakurai allylation反应立体选择性构建 syn-C5/C6 立体化学;(2)Mukaiyama aldol-lactonization 连续反应构建C2位多样性的不饱和六元内酯环。与已知路线相比,该路线的合成效率大幅提升:最长线性步骤由17步缩减至12步,总步骤从33步精简至24步,总收率由4%提升至19.7%,并在此基础上单批次制备了1.23 g 的Plocabulin。与秦勇和陈其凤团队进一步的合作研究表明,C2位取代基变化基本不影响其生物活性,这为未来开展ADC药物的研究提供了新的思路。
相关成果以发表在Org. Lett. 2025, 27, 4788-4793.(https://doi.org/10.1021/acs.orglett.5c01284)。研究得到了国家自然科学基金创新群体项目、国家重点研发计划等基金的资助,由我院博士生刘顺发,硕士生黎彬和高欣等共同完成。
进展三:海洋天然产物PM050463的首次和无保护基全合成
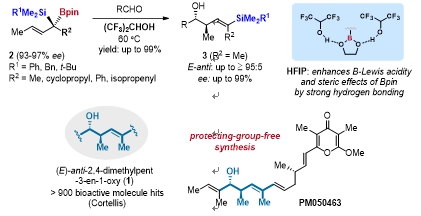
(E)-anti-2,4-dimethylpent-3-en-1-oxy广泛存在于聚酮天然产物中,Cortellis检索超过900个生物活性分子含有该共性结构单元。因此,但是,开发针对此类结构的高效和普适合成方法对于快速和多样性合成上述活性分子,拓展药学研究的深度和广度具有重要意义。
对此,该团队发展了新型的手性季碳型α-硅基巴豆基硼酸酯试剂,并发现六氟异丙醇(HFIP)作为溶剂不仅能增强该试剂巴豆基硼化的反应活性,还能生成此前难以获得的(E)-选择性。与四川大学化学学院苏志珊教授合作的分子动力学模拟表明,Bpin的氧原子与HFIP的质子间存在强烈的氢键相互作用,这增强了硼中心的Lewis酸性从而促进巴豆基硼化反应。通过DFT计算,不利的(Z)-anti-过渡态中HFIP与硅基间存在显著的空间排斥作用,最终导致了所观察到的不同寻常的(E)-anti-选择性。
该方法随后作为关键步骤被应用于海洋聚酮PM050463的合成中。PM050463提取自海洋来源的白色链霉菌。研究表明,其对由EGF-诱导的AP1激活有着强效抑制作用(IC50 <7.0 nM),有望开发成为新的抗癌药物。但是,该分子海洋提取困难且收率低,目前尚无合成报道。该团队基于上述方法学,实现了该分子的首次全合成,最长线性步骤6步,总步骤12步,且全程未使用保护基。
相关成果发表在Angew. Chem. Int. Ed. 2025, e202508944 (https://doi.org/10.1002/anie.202508944)。研究得到了国家自然科学基金创新群体项目、国家重点研发计划等基金的资助,由我院博士生童瑞棋、研究生刘诗阳等共同完成。
